

1. Introduction
The development of cost-effective, durable, non-toxic antimi- crobial systems, and highly efficient in different applications, such as in health products, dressings, hygienic materials, antimicrobial filters, coatings for medical equipment, textile production materi- als, among others, is of great interest nowadays [1,2]. For instance, the design of antimicrobial membranes based on colloidal polymer modified with multiwall carbon nanotubes decorated with silver nanoparticles [3] and membranes based on sulfonated polymer grafting and silver nanoparticles [4] have a promising application as an antimicrobial filtration membrane for water treatment. The results showed good bactericidal properties and suitable stability based on filtration test. On the other hand, membranes based on natural polymers and silver nanoparticles have potential applica- tion to be used as wound dressing [5,6].
Silver nanoparticles (AgNPs) have been used in several applica- tions due to their antifungal, antiviral, and antibacterial properties [7,8]. AgNPs are nanomaterials well known for their antibacterial properties, especially against Gram-negative (E. coli) and Gram- positive (S. aureus) bacteria, although being little susceptible to the development of bacterial resistance [9–11]. However, the silver toxicity and its environmental impact limit the use of pure AgNPs as antimicrobial agents. Thus, to overcome these drawbacks, a viable solution is to incorporate AgNPs into stable matrices, such as cellulosic substrates [12]. Cellulose, one of the most abundant natural biopolymers, [13], is obtained from a variety of organisms and has applications in various fields such as paper production, textile industries, and biomedical applications. In biomedical application, cellulose can be used in grafts and scaffolds in tissue engineering, skin dressings, drug delivery systems, among several others [14]. Therefore, the development of cellulosic materials pre- senting biological activities (antibacterial, antiviral, etc.) is extre- mely convenient. Thus, the development of techniques to induce antibacterial properties to cellulose against many types of patho- genic bacteria that affect the human health is required [15].
Cellulose nanowhiskers (CWs) can be obtained from the acid hydrolysis of cellulose, in which partial removal of the amorphous phase of the cellulose occurs maintaining the crystalline segments due to the differential hydrolysis kinetics of amorphous and crys- talline regions [16]. CWs have high surface area and high resistance as well as chemically reactive surface [17], with great potential in several applications such as controlled drug release, tissue engi- neering, functional reinforcement, and as a matrix for the synthesis of metallic nanoparticles, e.g. gold, platinum, and silver, that pre- sent antibacterial activity.
A common in situ method for AgNPs synthesis on fibrous sub- strates is based on the immersion of cellulosic materials into silver salt solutions followed by Ag+ reduction [1,18]. This immobiliza- tion process mitigates the nanoparticles aggregation. The stability of metallic nanoparticles can also be achieved by steric hindrance by attaching large molecules (capping agents) at their neutral surfaces controlling the approximation of the nanoparticles. Polyvinylpyrrolidone (PVP) [19], polyvinyl alcohol (PVA) [20], and gelatin [21], among other polymer/biopolymers, have been suc- cessfully used as capping agent for AgNPs.
Modification of the CWs also allows the stabilization of AgNPs. Succinic anhydride-modified CWs allowed the reactive sites (–COOH) to anchor and stabilize silver nanoparticles on CWs surfaces [22,23].
Drogat et al. (2011) [24] studied the formation of AgNPs by using CWs oxidized with periodate to generate aldehyde groups, which were used to reduce Ag+ under alkaline conditions. Antibac- terial assays were performed with E. coli and S. aureus showing inhibition effects. Jung et al. (2009) [25] reported AgNPs incorporated into cellulose membranes by the chemical reduction method using silver nitrate and sodium borohydride. The antimicrobial activity was more than 99.99% against E. coli and S. aureus.
Recently, the incorporation of metallic nanoparticles into poly- mer nanofibers has attracted attention, due to the high surface/vol- ume ratio, flexibility, and functionality of the surface of nanofibers [26], where they can be easily anchored providing distinct optical, electronic, catalytic, and antimicrobial properties [27]. Hong et al. (2006) [28] prepared nanofiber containing Ag by electrospinning of PVA/silver nitrate through temperature control and UV irradia- tion. The average diameter of Ag nanoparticles was 6.3 nm. PVA nanofibers containing Ag nanoparticles showed strong antimicro- bial activity against S. aureus (Gram-positive) and Klebsiella pneu- moniae (Gram-negative) bacteria. The samples were able to reduce ca. 99.9% of bacteria after 18 h of incubation.
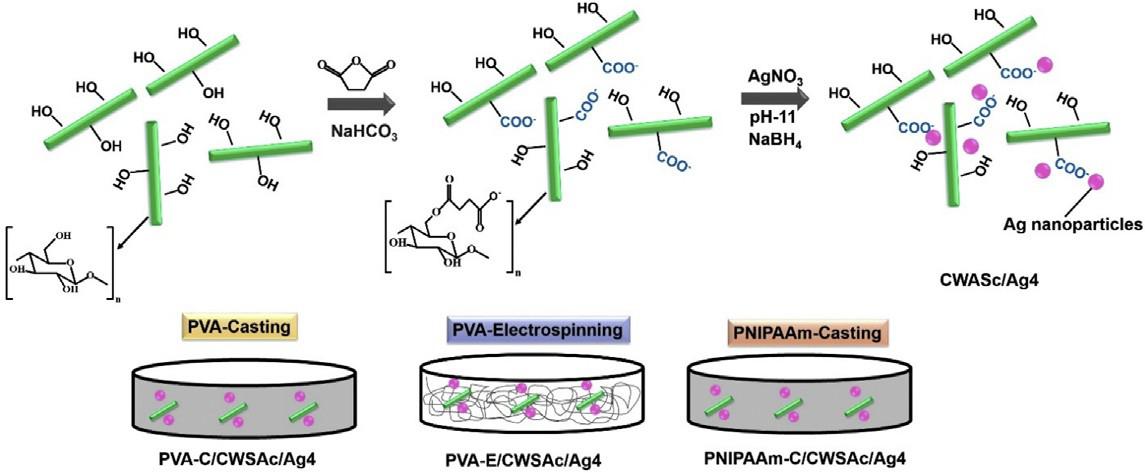
The use of CWs decorated with silver nanoparticles for employ- ing as additives to antibacterial polymer membranes can be inter- esting for several areas including membrane filtration, wound dressing, packaging among others. In this report, a simple method- ology without using organic solvents was developed to functional- ize CWs with carboxylate groups (–COO—), which were further successfully employed to anchor and stabilize silver nanoparticles (CWSAc/AgNPs). CWSAc/AgNPs were incorporated into polymer matrices of PVA (obtained by the casting and electrospinning method) and PNIPAAm (obtained by the casting method). All films with silver nanoparticles presented antimicrobial action against Gram-positive (S. aureus), and Gram-negative (E. coli andP. aeruginosa) bacteria. Therefore, the materials were proposed to address a long list of demands such as wound dressing, decontam- ination of drinking water, packaging, and for other cases where the proliferation of microorganisms are necessary. The present work explores the synthesis of the silver nanoparticles and its incorpora- tion into different polymer membranes prepared by electrospin- ning and casting. This study evaluates the suitable potential of the different membranes configuration against the proliferation of microorganisms.
2. Materials and methods
2.1. Materials
Cotton fibers were obtained from Cocamar (Agroindustrial Cooperative of Maringá, Maringá-PR, Brazil); glycerol (99%), poly (N-Isopropylacrylamide) (Mw 8.7 105 g mol—1), poly(vinyl alco- hol) 87–89% hydrolyzed (Mw 13.000 to 23.000 g mol—1), poly(vinyl alcohol) 88% hydrolyzed (88.000 g mol—1), silver nitrate, sodium borohydride and succinic anhydride were all obtained from Sigma-Aldrich. Hydrochloric acid (HCl-37%) and sodium bicarbon- ate were acquired from F. Maia (Brazil). Sodium hydroxide was obtained from Vetec (Brazil), and N, N-dimethylacetamide (DMA) was obtained from Nuclear (Brazil). All reagents were used as received without further purification.
2.2. Cellulose nanowhiskers
The synthesis of CWs was performed following the same proce- dure described by Spagnol et al. (2018) [29]. For this, cellulose-rich cotton fibers (2 g) were immersed in a NaOH solution (2% w/v) and kept under magnetic stirring for 1 h. The cotton was washed with excess of distilled water to removed the NaOH. The resulting cotton fibers were dried in an oven at 60 °C until constant weight. To obtain the CWs, 1 g of purified cotton fibers were hydrolyzed using concentrated HCl (20 mL, 37%) at 45 °C for 1 h, under magnetic stirring. The resulting suspension was centrifuged (10.000 rpm) for 5 min and washed several times with distilled water to remove the excess of acid (final pH 6–7). The final mate- rial was frozen and lyophilized.
2.3. CWs surface modification with succinic anhydride
The surface modification of CWs was performed following the same procedure described by Spagnol et al. (2018) [29], where 8 g of succinic anhydride (SA) were added to a one neck round bottom flask (50 mL) and kept at 130 °C until complete melting. 1 g of the as-prepared CWs was added and the medium was allowed to react for 24 h under magnetic stirring. Then, 20 mL of dimethylac- etamide (DMA) was added to the mixture and stirred for 20 min so the unreacted anhydride dissolved and could be removed from the reaction medium. It was filtered, washed with distilled water, and dried at 110 °C for 24 h. The modified-CWs were labeled as CWSA.
2.4. CWSA deprotonation
The deprotonation of the carboxylic groups from CWSA was achieved by adding 2 g of CWSA to saturated aqueous sodium bicarbonate solution (150 mL). The mixture was stirred for 30 min, then filtered, washed with distilled water and dried at 110°C for 24 h. The material was named as CWSAc.
2.5. Silver nanoparticles incorporation to CWSAc
CWSAc and AgNO3 aqueous solution was added to a beaker and kept under stirring for 15 h. Then, CWSAc impregnated with Ag+ ions was filtered and washed with an excess of distilled water. Subsequently, gelatin aqueous solution or distilled water, as well as a few drops of NaOH aqueous solution, were added to reach final pH 10–11. The mixture was stirred for 15 min, then, the NaBH4 aqueous solution was added and the medium was allowed to react for 24 h under magnetic stirring. It was filtered, washed with distilled water, and dried at 110 °C for 24 h. The samples were labeled according to the number of reactants used, as presented in Table 1.
2.6. PVA nanocomposite films
2.6.1. Casting films
PVA nanocomposite casting films were prepared by adding 8 w/w % of glycerol (relative to PVA mass) to 15 mL of 60 g L—1 PVA (Mw 13000–23000 g mol—1, 88% hydrolyzed) solution and stirred for 10 min. After 30 w/w% of CWSAc/Ag4 (relative to PVA mass) were added and the system was stirred for 15 min, sonicated for 2 min on a digital sonicator and transferred to a Teflon plate mold which was placed in a circulating air oven at 35 °C for 24 h.
The nanocomposite film was named as PVA-C/CWSAc/Ag4 (films obtained by using casting method with PVA and CWSAc/Ag4). PVA films without the addition of CWSAc/Ag were used as a control and labeled as PVA-C (films obtained by using casting method with PVA).
2.6.2. Electrospun films
PVA nanocomposite electrospun films were prepared with 10 mL of 60 g·L—1 PVA (Mw 13000–23000 g·mol—1, 88% hydrolyzed) and 30 w/w% of CWSAc/Ag4 (relative to PVA mass). The system was stirred for 15 min and sonicated for 5 min on a digital sonica- tor and taken to electrospun. The nanocomposite film was named as PVA E/CWSAc/Ag4 (films obtained by using electrospun method with PVA and CWSAc/Ag4). PVA electrospun fibers, named as PVA- E (films obtained by using electrospun method with PVA), were used as a control.
Electrospinning was performed at room temperature using PVA solutions. The samples were electrospun at 15 kV, flow rate of 0.3 mL/h and distance between the needle tip and the collector was 15 cm.
Label | CWSAc | AgNO3 solution | Gelatin solution (0.2% m/v) | Distilled water | NaBH4 (0.1 M) |
|---|---|---|---|---|---|
CWSAc/Ag1 | 0.5 g | 30 mL (0.05 M) | 15 mL | 0 | 0.4 mL |
CWSAc/Ag2 | 0.5 g | 20 mL (0.025 M) | 0 | 0 | 0.1 mL |
CWSAc/Ag3 | 0.5 g | 30 mL (0.05 M) | 0 | 15 mL | 0.6 mL |
| CWSAc/Ag4 | 0.5 g | 30 mL (0.05 M) | 0 | 15 mL | 0.4 mL |
2.7. Pnipaam nanocomposite films
PNIPAAm nanocomposite films were obtained by using casting method as follows: 15 mL of 8 w/v% solution of PNIPAAm in etha -nol was prepared. After 10 min of stirring, 30 w/w% of CWSAc/Ag4 (relative to PNIPAAm mass) were added and the medium was allowed to react for 15 min under magnetic stirring. Then the solution was sonicated for 2 min and transferred to a teflon mold, which was placed in a circulating air oven at 35 °C for 24 h. The nanocomposite films were named as PNIPAAm-C/CWSAc/Ag4 (films obtained by using casting method with PNIPAAm and CWSAc/Ag4). As a control sample, PNIPAAm films were prepared without addition of CWSAc/Ag4, and they were named as PNIPAAm-C (films obtained by using casting method with PNIPAAm).
2.8. Characterization
2.8.1. Scanning electron microscopy (SEM) coupled with X-ray dispersive energy spectroscopy (EDS)
SEM measurements were performed with a scanning electron microscope coupled with X-ray dispersive energy spectroscopy, Shimadzu, SSX-550 Superscan model. The accelerating voltage was 15 kV and electric current was 30 mA. The surfaces of the sam- ples were previously coated with a thin layer of gold by sputtering technique. The chemical mapping was performed in the same apparatus where the colors were defined for the elemental analysis.
2.8.2. Transmission electron microscopy (TEM)
A suspension of CWSAc/AgNPs was prepared in deionized water and placed on ultrafine supports (grids) of carbon-coated copper (400 mesh). The analysis was performed by a TOPCON 002B trans- mission electron microscope by using an acceleration voltage of 200 kV.
2.8.3. Thermogravimetry (TGA)
TGA was performed on samples from 25 to 1000 °C, at a heating rate of 10 °C/min under 50 mL/min of flowing nitrogen atmosphere in a thermogravimetric analyzer (Shimadzu, TGA 50).
2.8.4. X-ray diffractometry (XRD)
The diffractograms of the nanocrystals were recorded in a Shimadzu diffractometer, XRD-7000 model, by using monochro- matic CuKa (= 1.5418 Å), the voltage was 40 kV and the current was 30 mA. The measurements were performed in continuous scan mode with a scan rate of 2° min—1 in the Bragg (2h) de 10° an 80°.
2.8.5. Attenuated total reflectance fourier transform infrared spectroscopy (ATR-FTIR)
ATR-FTIR- was performed on a BOMEM spectrometer, MB-100 model, accumulating 32 scans in the range of 4000– to 630 cm—1, with 4 cm—1 of resolution.
2.9. Application
The antimicrobial activity of the CWSAc/AgNPs and nanocom- posite films was evaluated against Staphylococcus aureus (S. aur- eus - ATCC25923), Bacillus subtilis (B. subtilis - ATCC 6623), Escherichia coli (E. coli - ATCC 25922) bacteria and Candida albi- cans fungus (C. albicans - ATCC 10231), following the procedure described by Follmann et al. (2016, 2018) [30,31] and Ruparelia et al. (2008) [32].
The bacteria were cultured in nutrient broth at 37 °C and stored on lean nutrient agar at 4 °C. The yeasts were cultivated and stocked on sloping Sabouraud agar.
2.9.1. Preparation of the inoculum
For each bacterial sample, a standard suspension was prepared from the 24 h culture in Mueller-Hinton broth (MH-broth). In a tube containing sterile saline (NaCl - 0.9%), the bacterial culture was added dropwise until a turbidity identical to that obtained with the 0.5 tube of the McFarland scale (corresponding to 1.5 108 Colony Forming Unit (CFU)/mL). Yeasts were standardized from the 48 h culture in Sabouraud broth. In another tube contain- ing sterile saline solution was added dropwise until turbidity was obtained, standardized according to the 0.5 tube of the McFarland scale (1.5x108 CFU/mL).
2.9.2. Disk diffusion test
The detection of antimicrobial activity of the silver nanoparti- cles powder incorporated in the CWs and of the nanocomposite films with the incorporation of CWASc/Ag4 against the bacteria tested were performed by the disc diffusion method (CLSI, 2009). Inoculum suspension (Section 2.9.1) was used to inoculated Muller-Hinton or Sabouraud broth agar plates by using a sterile swab. Then, CWASc/Ag (10 mg) were placed on the plates equidis- tant on the surface of the medium with the aid of a sterile glass disc (which was subsequently removed). For the nanocomposite films, 4 disks of the films (diameter 7 mm) were placed aseptically, equidistant, on the surface of the medium. Incubate at 37 °C for 24 h. The antibacterial activity was evaluated by the formation of inhibition halos, which were measured and expressed in millime- ters. The assays were performed twice for each sample.
2.9.3. Determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC)
The inoculum suspensions were diluted (1:10) in MH-broth medium to obtain a bacterial suspension of 1.5 107 CFU/mL to perform the minimum inhibitory concentration (MIC) assays. Briefly, to determine the MIC, 20 mg/mL stock solutions of CWSAc/Ag were dispersed in 10% solutions of DMSO. Subse- quently, a 1:10 dilution was performed in MH-broth and another with Sabouraud broth. Using a 96-well microplate, 100 lL culture medium (MH-broth or Sabouraud broth) was added to all wells.
Then 100 lL of stock solutions (1:10) were pipetted into the first well, in which an initial concentration of 1000 lg/mL was obtained. A serial dilution in the range of 1000 lg/mL to 15.6 mg/ mL was performed from the first well, homogenizing and transfer- ring 100 lL from the first well to the second well, from the second well to the third well, and so on up to the seventh well. Volumes of 5 lL of the bacterial suspension prepared as described above (107 CFU/mL) or yeasts (107 CFU/mL) were added to each well. The microplates were incubated for the bacteria in a humid camera at 37 °C for 24 h and for the yeasts in a humid camera at 37 °C for 48 h. The tubes were then examined by visible turbidity of the solutions. The MIC was defined as the highest dilution where there was inhibition of growth, i.e. absence of visible turbidity. To determine the MBC of CWSAc/Ag, 10 lL of culture solution was added to individual wells in the culture plate from the highest to the lowest concentration of silver nanoparticles incorporated in the CWs able to inhibit microbial growth on plates of petri contain- ing the bacteria in MH broth or yeast in Sabouraud broth. The microplates were incubated at 37 °C for 24 h. The reading was verified through the verification of the growth, by the formation of colonies. MBC was defined as the lowest concentration of each sample that totally prevented the microbial growth in the plates. The test were performed three times for each sample.
3. Results and discussion
Purified cotton fibers were obtained by removing proteins (2% w/v NaOH, 2 h), waxes and other minor impurities with extensive rinsing of crude cotton. Acid hydrolysis (37% HCl, 20 mL/g, 45 °C, 1h) of pure cotton fibers yielded 75% cellulose nanowhiskers (CWs). CWs displayed a white fine powder appearance before modifica- tion turning to brownish-color after modified with succinic anhydride (insertion of –COOH groups). TEM images revealed CWs, CWSA and CWSAc to be comprised of nanometric needle-shaped structures with average lengths of 221.4 ± 61.30 nm, 69.40 ± 19.8 0 nm and 67.60 ± 19.80 nm, respectively, Fig. S1 in the supporting information. CWs were slightly grayish after the incorporation of AgNPs.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jcis.2018.07.096.
Aschematic route used in the preparation of either casting or electrospun nanocomposite films is presented in Fig. 1. CWs were functionalized with carboxylate groups that acted as anchoring sites for Ag+ ions via electrostatic interactions allowing stabiliza- tion of silver ions and their uniform distribution throughout the CWs surfaces. CWSAc/Ag+ were converted to CWSAc/Ag by sponta- neously reducing Ag+ ions to Ag0 catalyzed by the alkaline medium and the presence of –OH groups on CWs [33,34]. The films obtained by casting and electrospinning presented a smooth, highly homogeneous appearance and in the case of electrospun PVA films, they were highly flexible. The coloration of the films after the addition of the CWSAc/AgNPs was gray.
CWSA/Ag1 morphology observed on SEM (Fig. 2a) was similar to that of CWSAc (Fig. S2 in the supporting information). TEM images revealed the presence of small and uniform AgNPs dis- tributed throughout the CWs surfaces, Fig. 2b, which was corrobo- rated by EDS spectrum, Fig. S3 in the supporting information, and SEM atomic mapping, Fig. 3. Note that, the smaller nanoparticles, as observed in the histogram with a mean size of 6.2 ± 3.0 nm (Fig. S4 in the supporting information), were mainly associated with the presence of the colloidal stabilizer (gelatin) in the synthesis of nanoparticles, which stabilized and avoided aggrega- tion and uncontrolled growth of AgNPs. Stabilization of AgNPs was due to the –COOH and NH2 groups on gelatin. The nonbonding electron pairs of the oxygen and nitrogen atoms can occupy two silver ions sp orbitals to form a complex. This coordination com- plex between silver ions and gelatin produced a protective col- loidal layer preventing the agglomeration of these particles [35,36]. However, CWSAc/Ag2, CWSAc/Ag3 and CWSAc/Ag4 mate- rials (Fig. 2(c), (e) and (g)) presented changes in their morphology, such as rough surfaces associated with the presence of silver, as confirmed by TEM images, Fig. 2d, f, g and EDS spectra (Fig. S3 in the supporting information).
The synthesis of CWSAc/Ag2, CWSAc/Ag3, and CWSAc/Ag4 without the use of a colloid protector generated larger particles with a population of particles and distribution differing signifi- cantly to that of CWSAc/Ag1. Silver particles became more numer- ous and even non-spherical, possibly due to the aggregation of smaller particles which were not visualized by SEM. Due to the
Fig. 2

Fig. 2
SEM (left side) and TEM (right side) images of CWSAc/Ag1 (a and b), CWSAc/ Ag2 (c and d), CWSAc/Ag3 (e and f) and CWSAc/Ag4 (g and h).
Fig. 3.

Fig. 3.
(a) SEM image and (b) silver atomic distribution mapping (CWSAc/Ag1 sample).